|
|
|
|
|
|
|
|
| Encefalopatias
espongiformes transmissíveis
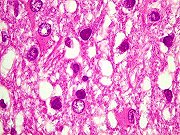
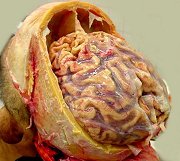
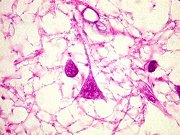
(doenças causadas por prions). Definição. São doenças associadas a formas anormais de uma proteína específica, chamada de proteína priônica ou príon (PrP), normalmente expressada em neurônios, mas de função ainda desconhecida. Fazem parte do grupo a doença de Creutzfeldt-Jakob (CJD), síndrome de Gerstmann-Sträussler-Scheinker (GSS), insônia familial fatal e o kuru. Em animais existem a doença chamada de scrapie em carneiros e cabras e a encefalopatia espongiforme bovina ou doença da vaca louca, entre outras. A scrapie é uma doença que causa prurido cutâneo nestes animais, levando-os a esfregar-se nas cercas causando ferimentos e perda do valor comercial das peles. Todas estas doenças são ao mesmo tempo infecciosas e transmissíveis geneticamente, e são caracterizadas morfologicamente pela chamada alteração espongiforme, que é o acúmulo de vacúolos intracelulares em neurônios e glia e, clinicamente, por demência progressiva. A apresentação mais comum é a CJD, cuja forma esporádica responde por cerca de 90% dos casos e tem incidência anual de 1 caso por milhão de pessoas. O resto é de formas familial e infecciosa. Patogênese e genética molecular. A proteína PrP é uma proteína normal de 30 kD presente em neurônios. A doença ocorre quando a proteína sofre uma alteração conformacional de sua isoforma normal que contém uma alfa-hélice a uma isoforma anormal contendo pregueamento beta. Esta isoforma, chamada PrPsc (sc para scrapie), é resistente a digestão por proteases. A alteração conformacional pode ocorrer espontaneamente a taxas extremamente baixas, resultando nos casos esporádicos, ou com freqüência mais alta quando certas mutações estão presentes na proteína PrP, como ocorre nas formas familiais das doenças priônicas. A proteína alterada PrPsc, quando presente, e independente de seu mecanismo de origem, promove a alteração conformacional correspondente nas moléculas vizinhas de proteína PrP normal. Esta é a base para a natureza infecciosa da doença. Uma vez introduzidas num animal normal, as proteínas alteradas modificam as proteínas existentes. Assim, material preparado a partir de casos humanos esporádicos ou familiais mostra ser infeccioso quando inoculado em hospedeiros susceptíveis. Se o animal tiver sido geneticamente modificado para não produzir a proteína PrP através de inativação do gene PRNP este animal será resistente à infecção (já que não terá proteínas normais para sofrerem a modificação patológica. O gene PRNP que codifica a proteína PrP está localizado no braço curto do cromossomo 20 e é altamente conservado entre espécies. Nos casos familiais das doenças priônicas, certas mutações pontuais foram identificadas no gene PRNP, que favorecem a ocorrência da doença ou de certos padrões patológicos. Como exemplo, o códon 129 é importante. A heterozigose a nível deste códon parece proteger da doença. Indivíduos que são homozigotos no códon 129 para metionina ou valina são proporcionalmente mais numerosos entre os casos de CJD. Os casos familiais têm padrão de herança autossômico dominante. Aparentemente, o acúmulo da proteína modificada no tecido nervoso seria responsável pelas alterações patológicas, mas como isto exatamente ocorre não é compreendido, em particular, como se formam os vacúolos intracitoplasmáticos da encefalopatia espongiforme. Principais formas das doenças priônicas. Doença de Creutzfeldt-Jakob (CJD). Esta doença rara manifesta-se por demência rapidamente progressiva com pico na 7ª. década. Reconhecem-se formas familiais e transmissão iatrogênica, por exemplo por transplantes de córnea, enxertos de dura-máter, implantação de eletrodos e injeção de hormônio de crescimento preparado de hipófises humanas. Clínica. O início é com alterações sutis de memória e comportamento, seguidos por rápida progressão a demência. É característica a ocorrência de mioclonias (contrações involuntárias bruscas dos músculos, espontâneas ou quando o paciente é estimulado, particularmente por ruídos altos startle myoclonus). Numa minoria dos pacientes há ataxia e outros sinais de disfunção cerebelar, e/ou alterações visuais e piramidais. As alterações visuais refletem afecção predominante dos lobos occipitais na chamada variante de Heidenhain. O padrão predominantemente cerebelar é chamado variante de Brownell-Oppenheimer. As alterações no EEG são características, com complexos periódicos de onda aguda seguida por uma onda lenta (complexos ponta-onda) com freqüência aproximada de 1 por segundo. A doença é sempre fatal com duração geralmente de alguns meses, mas a sobrevida pode ser maior. Patologia. A alteração principal e mais característica é a formação de pequenos vacúolos aparentemente vazios no neurópilo e, às vezes, no pericário de neurônios (encefalopatia espongiforme). Em microscopia eletrônica, os vacúolos são limitados por membranas, e parecem estar no interior de prolongamentos neuronais. Há também perda neuronal severa e gliose reacional, com redução no volume das estruturas afetadas, principalmente córtex cerebral, núcleos da base e córtex cerebelar. Isto resulta macroscopicamente em atrofia, que pode ser muito intensa. Contudo, em parte dos casos, não há atrofia macroscópica, devido à rapidez da doença. Nos casos avançados, a identificação da encefalopatia espongiforme pode ser difícil devido à extensa destruição tecidual. Neste caso fala-se em status spongiosus. Tipicamente, não se observa infiltrado inflamatório. Outra alteração característica são as placas do tipo kuru, que são depósitos extracelulares de proteína anormal com as características de amilóide. São positivas para vermelho do Congo e PAS-positivas. São abundantes no córtex cerebral nos casos de doença de Creutzfeldt-Jakob variante (abaixo) e no cerebelo nos casos de síndrome de Gerstmann-Sträussler-Scheinker (abaixo). Imunohistoquímica demonstra nestas estruturas a proteína PrPsc anormalmente agregada e resistente a proteases. Doença de Creutzfeldt-Jakob variante (vCJD). Esta forma iniciou-se em 1995 no Reino Unido como casos que diferiam da CJD clássica por afetar adultos jovens, com alterações de comportamento proeminentes, e progressão mais lenta da síndrome neurológica. Muitos pacientes não apresentavam as alterações no EEG típicas da CJD. Patologicamente as alterações são muito semelhantes às da CJD clássica, mas com grande proeminência de placas do tipo kuru circundadas por alteração espongiforme. Não havia alteração do gene PRNP, mas todos os pacientes eram homozigotos para metionina no códon 129. Várias evidências ligaram esta variante à encefalopatia espongiforme bovina ou doença da vaca louca (BSE), que teria sido transmitida a humanos por injestão de carne contaminada. Atualmente, admite-se que a epidemia ficou limitada a algumas centenas ou milhares de casos. Síndrome de Gerstmann-Sträussler-Scheinker (GSS). Doença autossômica dominante herdada com mutações do gene PRNP, que tipicamente começa com ataxia cerebelar, disartria e nistagmo e se continua com demência, mas esta é relativamente leve. O curso é mais lento que a CJD e pode durar vários anos. Além da encefalopatia espongiforme, a doença apresenta placas de amilóide contendo PrP, e alterações neurofibrilares. A insônia familial fatal (FFI), também autossômica dominante, é assim chamada devido às alterações do sono (insônia intratável) nos seus estágios iniciais. Depois aparecem ataxia, distúrbios autonômicos (hiperatividade simpática), distúrbios endócrinos, estupor e coma. A duração é de cerca de 3 anos. As alterações são perda neuronal e gliose em núcleos mediais dos tálamos e nos núcleos olivares inferiores, sem alteração espongiforme. Kuru. A palavra quer dizer tremor na língua Fore dos nativos da Nova Guiné, onde era prevalente e foi inicialmente observada. Foi a primeira infecção lenta descrita em seres humanos. A doença se manifesta como uma ataxia cerebelar progressiva, com anormalidades dos movimentos extraoculares, fraqueza progredindo a paralisias, incontinência esfincteriana a e morte em 3 6 meses. Foram notadas importantes semelhanças epidemiológicas e patológicas entre o kuru e o scrapie (1959), sugerindo que o kuru pudesse ser transmissível a primatas, o que se obteve em 1966. Inoculação de material cerebral de pacientes com kuru em chimpanzés resultou em doença após latência de 18 a 36 meses. Material destes passou a doença a outros chimpanzés. Kuru era transmitido entre os nativos através de canibalismo ritual, em que os tecidos do morto eram ingeridos e esfregados na pele dos parentes, principalmente as mulheres e crianças de ambos sexos. Com isso havia absorção do agente infeccioso por ferimentos na pele, conjuntivas e outras mucosas. A abolição da prática levou ao gradual desaparecimento da doença. Precaução na manipulação de espécimes em casos suspeitos de CJD. Como o agente não é um vírus, não contém ácidos nucléicos e não é inativado por fixação formólica convencional. Recomenda-se, após fixação em formol a 10% por 24 horas, imersão dos tecidos em ácido fórmico a 100% por pelo menos uma hora, retornando então o espécime ao formol até o processamento para inclusão em parafina. Todos materiais não descartáveis, inclusive instrumental cirúrgico ou de autópsia, que tenham tido contato com tecidos, devem ser imersos em hidróxido de sódio 2N (80 g de NaOH por litro de água) por pelo menos uma hora. Extraído na maior parte de Frosch MP, Anthony DC, De Girolami U. The Central Nervous System. in Kumar V, Abbas AK, Fausto N (eds). Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Elsevier Saunders, Philadelphia, 2005. pp. 1380- 2. com informações adicionais de
|
| Para página de CJD na graduação e mais imagens, clique » |
 |
 |
 |
| Módulo Neuro - Página Inicial | Outros módulos | e-mails : [email protected]___[email protected] |